
African Journal of Respiratory Medicine received 855 citations as per google scholar report
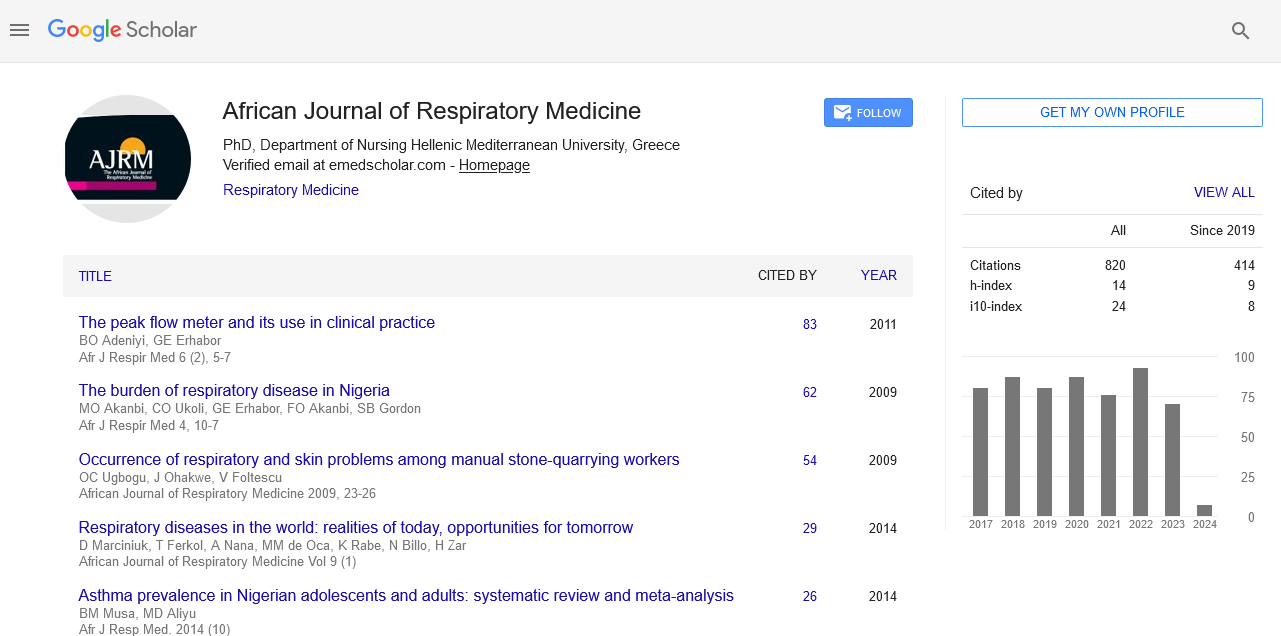
Case Report - (2020) Volume 15, Issue 1
Received: 18-Aug-2020 Accepted Date: Aug 27, 2020 ; Published: 31-Aug-2020
Inflammatory myofibroblastic tumor is an uncommon benign neoplasm, which frequently involves the lung in children and young adults; it has the tendency to be locally invasive and recurrent but rare metastasis. We present the case of a 40-year- old Sudanese female with pleural inflammatory myofibroblastic tumor invading adjacent chest wall and presenting with nonspecific respiratory symptoms and signs.
Inflammatory myofibroblastic tumor (IMT), or plasma cell granuloma, is a rare benign tumor that was first described by Bahadori and Liebow.1 IMT frequently involves the lungs, abdominopelvic region and maxillofacial structures. The tumor mainly occurs in children and young adults.2 Patients usually present with a productive cough and generally lesions are benign, with recurrence, and local blood vessels invasions, however metastasis is rare.3 Pulmonary IMT most commonly presents as a solitary thoracic nodule or clear mass on chest radiography,4, 5 and rarely presents in the pleura.6 Histology reveals the proliferation of spindle cells with a prominent inflammatory infiltrate consisting of plasma cells and lymphocytes, with occasional eosinophils and neutrophils.7 A definitive diagnosis can be made with the immunohistochemical stain.8 In this paper, we present the case of a 40-year- old Sudanese female with pleural IMT invading adjacent chest wall and presenting with nonspecific respiratory symptoms and signs.
We present a 40-year- old female who presented with symptoms that had been evolving over 3 months, including cough and right chest pain. She has no associated shortness of breath or fever and

Figure 1: (A) Chest radiography revealing opacity on the right upper lung zone on the right side. (B) Computed tomography confirming a peripheral mass in the upper lobe of the right lung, with local invasion and destruction of the second rib.

Figure 2: (A) Histological examination (hematoxylin-eosin staining) shows spindle cell proliferation with the infiltration of lymphocytes and plasma cells. (B) Immunohistochemically, neoplastic cells are focally positive for smooth muscle actin.
Histopathology sections showed numerous plasma cells with adjoining bone trabeculae, inflamed fibrous connective tissue, and striated muscle fibers (Fig. 2A). In immunohistochemistry LCA, CD3, Kappa, and Lambda were positive, but CD20 was negative (Fig. 2B). The immunohistochemistry suggested IMT and the patient was referred for thoracic surgical resection of the mass and any further management if required.
IMT is a rare tumor that frequently involves the lung, and is found mainly in children and young adults. It tends to be locally invasive or recurrent and rarely metastasizes. The etiology and pathogenesis are unknown. Several theories suggested immunologic etiology. Griffin et al. reported findings suggesting that IMT is a neoplastic rather than reactive subset. Patients with IMT usually present with non-specific pulmonary features,3 and their chest imaging usually reveals solitary pulmonary nodules.4, 5 As in our case report, histopathology sections reveal the proliferation of spindle cells with a prominent inflammatory infiltrate consisting of plasma cells and lymphocytes, with occasional eosinophils and neutrophils which highly suggestive of IMT.7 Immunohistochemistry markers are not specific but can support the diagnosis.8 The primary treatment is surgical resection; but if not respectable and/or metastatic inflammatory myofibroblastic tumor can be treated with systemic therapies, including glucocorticoids, radiotherapy, and/or chemotherapy. In our case, symptoms were nonspecific; radiology suggested invasive tumor and immunohistochemistry confirmed the diagnosis of IMT, and the patient was referred for surgical treatment.
IMT is rare, but frequently involves the lung, and should be in the list of the differential diagnosis for lung masses. Complete surgical removal is the primary treatment. However, when surgical excision is not possible and the metastatic inflammatory myofibroblastic tumor can be treated with systemic therapy, including glucocorticoids, radiotherapy, and/or chemotherapy.
Written informed consent for publishing the case details and images were taken from the patient and the patient’s family and an IRB exemption was obtained from the Hospital ethical committee.
All authors declare that they have no conflicts of interest in all steps of this case report.
The authors are thankful to the management of Al-Maarefa University, Riyadh, Kingdom of Saudi Arabia, for providing necessary facilities to carry out and publish this work.
Select your language of interest to view the total content in your interested language
To read the issue click on a cover

